- ESM-2 — for generic proteins. Use it for embeddings or per-position mutation likelihoods on enzymes, receptors, scaffolds, or any non-antibody protein.
- AbLang-2 — for paired antibodies. Use it on heavy + light chain pairs for antibody-aware embeddings or per-position mutation likelihoods that reflect natural antibody repertoires.
What each capability does
Embeddings
An embedding is a fixed-length vector that represents a full protein sequence. Two sequences with similar embeddings are treated as similar by the underlying model, typically reflecting similar fold, family, or function. Use embeddings when you want to:- Cluster a set of sequences by similarity
- Compare two or more proteins numerically
- Search a library for sequences similar to a reference
- Build features for downstream ML (activity prediction, property prediction)
Mutation likelihoods
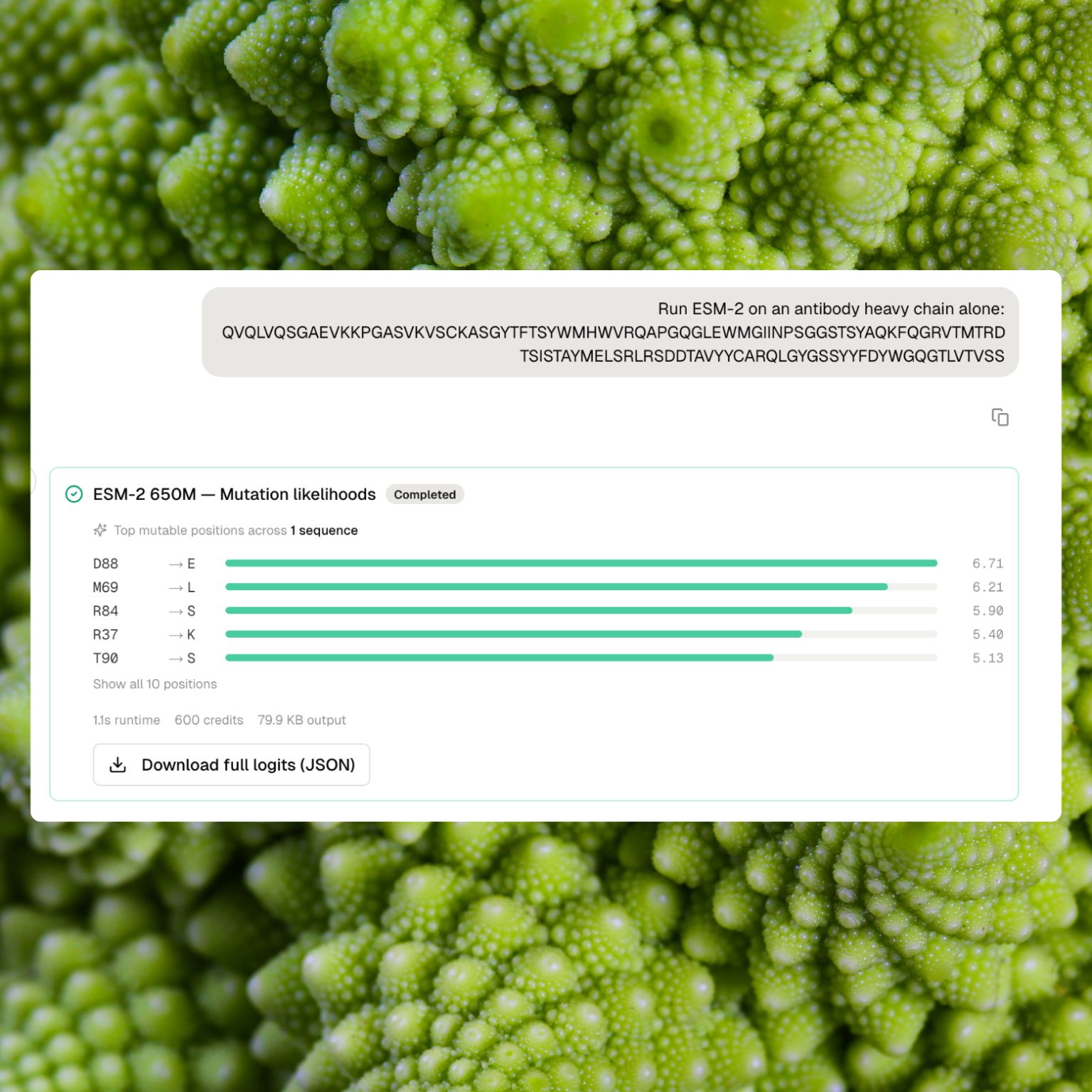
A mutation likelihood is a per-position score for every possible amino acid substitution at every residue. High scores at a position mean the model finds multiple alternatives plausible, which is often a signal of an evolvable, tolerant, or flexible site. Low scores mean the model strongly prefers the natural residue. Use mutation likelihoods when you want to:- Identify candidate sites for mutagenesis or affinity maturation
- Flag unusual residues in a sequence
- Rank substitutions at a known hotspot
- Get a starting point for protein engineering, before committing to wet-lab rounds
When to use ESM-2 vs AbLang-2
AbLang-2 is trained on paired antibody repertoires, so its likelihoods and embeddings reflect the statistics of real antibody sequences — CDR variability, framework conservation, and the pairing between heavy and light chains. ESM-2 is a general-protein model: broader coverage, no antibody-specific bias.
For antibodies, start with AbLang-2 for repertoire-aware signal. For everything else, start with ESM-2.
Asking for a run
You can trigger either capability with a natural-language prompt. A few examples: ESM-2 embeddings- “Compute ESM-2 embeddings for this sequence: MKWV…”
- “Encode these 5 protein sequences with ESM-2 so I can cluster them.”
- “Give me per-position mutation likelihoods for this enzyme using ESM-2.”
- “Run ESM-2 mutation scoring on this protein and show me the top mutable sites.”
- “Encode these heavy/light chain pairs with AbLang-2.”
- “Give me AbLang-2 embeddings for this antibody pair.”
- “Run AbLang-2 mutation likelihoods on this paired antibody.”
- “Show me the top mutable CDR positions for this antibody pair.”
Input formats
ESM-2
Paste sequences directly in the message, or attach a FASTA file.
AbLang-2
Provide the heavy and light chain for each antibody. MIP uppercases sequences and strips whitespace automatically.
The result card
When you submit, an inline card appears in the chat. It updates in place without a page refresh. While running: a compact status card shows the model, the action, and a spinner. You can continue the chat — the card tracks its own job. When complete:- Embeddings runs show the number of sequences encoded and the vector dimensionality (for example,
3 × 1280 dimsfor ESM-2 or2 × 480 dimsfor AbLang-2), plus a one-line hint on common uses. - Mutation-likelihood runs show a ranked list of the top mutable positions with their highest-likelihood substitutions and scores. Positions are labeled with the observed residue and a clear arrow to the predicted alternative (for example,
Q127 → E (3.02)). The list starts with the top 5 and expands on request.
Downloading the full output
Both capabilities save their full numerical output as a downloadable dataset, accessible from:- The result card’s Download action
- The Files view, under your datasets
Pricing and limits
Most runs finish in under a minute. A single billed minute is the typical cost for a small batch.
Enabling models in your workspace
Protein embeddings and mutation likelihoods are managed per model in your capability settings, so you can turn on exactly the ones you need:- AbLang-2 — Embeddings
- AbLang-2 — Mutation likelihoods
- ESM-2 — Embeddings
- ESM-2 — Mutation likelihoods
When to use which in a single workflow
A common pattern is combining the two model families across a multi-step analysis:- Embed a batch of candidates to cluster them and pick representatives.
- Score mutation likelihoods on the representatives to find tolerant positions.
- Pick a small set of mutations at the top-scoring positions for wet-lab testing.
Both ESM-2 and AbLang-2 are computational models. The outputs are ranking signals and feature representations, not experimental measurements. Treat them as hypothesis-generating inputs to your downstream workflow.